



内容
DIMDI全称:German Institute of Medical Documentation and Info
中文翻译:德国卫生部
DIMDI备案流程:
1. 大致审核资料(产品+包装图片、说明书以及公司信息等)和证书。
2. 大致沟通关于备案的相关流程和细节,待客户确认后即开始备案。
与此同时,安全官针对客户的产品、包装和说明书提出修改建议,客户同意后将修改后的包装、说明书和产品样品寄给我司(10pcs)。注:修改建议仅为基础建议(欧代+制造商信息,CE标,其他非必要的Logo和标记都不要出现),只为通过药监局审核,不构成任何合规建议或承诺。
3. 由客户指定的技术员对产品进行一次视频介绍,主要内容包括产品的功能讲解、产品使用注意事项、使用中可能遇到的突发状况以及应对办法。
4. 客户同期准备产品技术文件,相应要求见附录。
5. 在收到药监局的证书和客户介绍视频及样品后出具欧代证书。
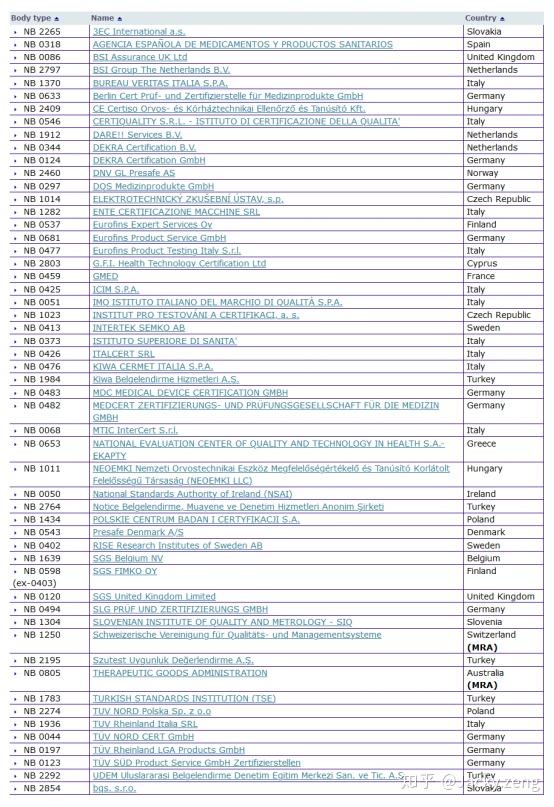
附录:CE证书认可机构列表。
MDD认证,即93/42/EEC指令认可的NB机构:
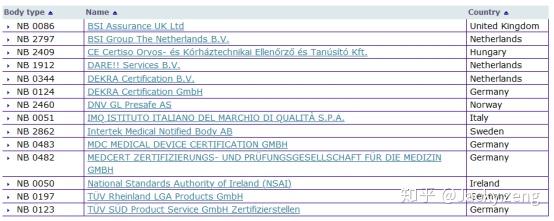
MDR认证,即EU 2017/745指令认可的NB机构:
技术文件要求:
亚马逊要求DIMDI注册
制造商必须拥有并保存各个级别医疗器械(I、IIa、IIb、III)的最新技术文档(也称作技术文件或器械主文件)。该文档用于证明相关医疗器械符合基本要求(93/42/EEC 附录 I)。技术文件的内容至少应包括:
・ 目录
・ 关于结构和用途的常规信息
・ EC 符合性声明及根据93/42/EEC 附录IX 的分类
・ 制造商/欧洲代表及制造工厂的名称和地址
・ 产品说明
・ 产品说明(包括所有变体)
・ 产品图片
・ 产品使用图片
・ 手册、广告、目录等(如有)
・ 产品规格
・ 适用标准列表
・ 部件列表,组件列表
・ 装配和/或制造图纸
・ 部件装配图纸、组件图纸(线路)
・ 使用材料规格(包括数据表)
・ 如果同时是药品器械:关于使用成分的说明(如适用,也应包含咨询程序结果)
・ 制造规格
・ 消毒规格
・ 包装规格
・ QA 规格(如QC 规格、流程内和最终控制、最终发布等)
・ 标记、随附文档、包装说明书(如EN 980、ISO 15223)
・ 批号设计说明
・ 使用说明(如EN 1041)
・ 预期/临床使用
・ 症状、禁忌症状
・ 操作说明、使用说明
・ 警告/注意事项
・ 服务手册
・ OEM 合同或供应商认证
・ 产品检验
・ 测试数据和报告、功能性研究、水下实验室或实验台测试
・ 材料认证/稳定性、生物测试和无菌室监测报告
・ EMC 测试和认证
・ 兼容性研究(与其他器械连接)
・ 风险管理文档(EN ISO 14971)
・ 临床数据(如EN 14155-1/-2,MEDDEV 2.7.1,第15 条/附录10)
・ 产品验证
・ 包装验证/老化研究
・ 流程验证(如消毒、制造、生产)
・ 软件验证
医疗器械DIMDI注册